Thomas Bosch Lab
The Bosch lab studies the intricate interactions within metaorganisms in a simple model system, the non-senescent cnidarian Hydra.


By loading the video, you agree to YouTube’s privacy policy.
Learn more
Understanding aging and how it affects an organism’s life span is a fundamental problem in biology.
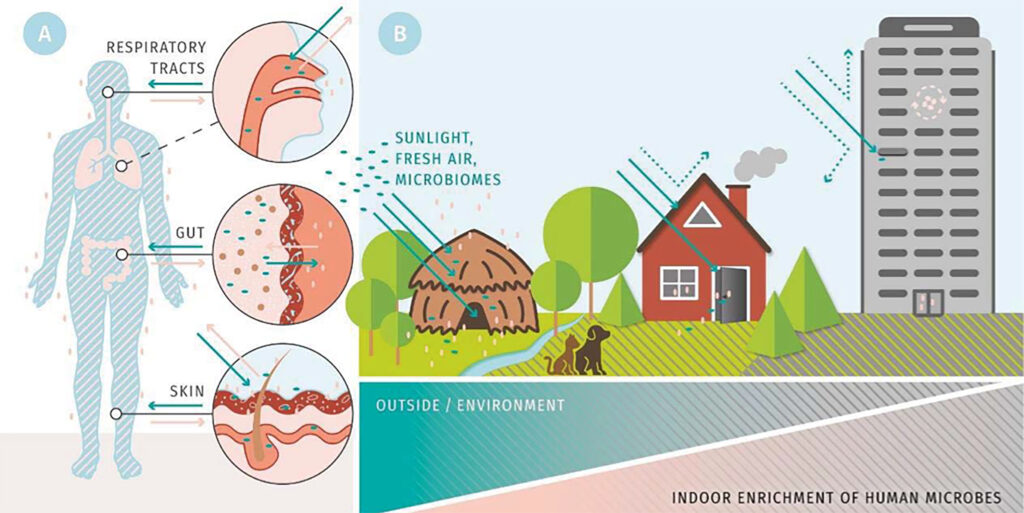
A hallmark of aging is stem cell senescence, resulting in an impaired regenerative capacity and reduced tissue function. In addition, aging is characterized by profound remodelling of the immune system, a phenomenon referred to as immune-senescence. Yet, what is causing stem cell and immune-senescence? Moreover, multicellular organisms exist as metaorganisms comprised of the macroscopic host and synergistic interdependence with numerous microbial and eukaryotic species. Health, therefore, is fundamental multi-organismal.
Realizing that animals exist only within a partnership with symbionts as metaorganisms has led to two important realizations:
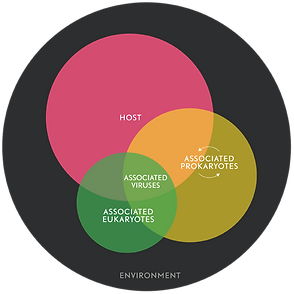
- First, it is becoming increasingly clear that to understand the physiology, evolution and development of a given species, we cannot study the species in isolation.
- Second, the health and fitness of animals, including humans, appears to be fundamentally multi-organismal. Disturbance within the complex community can have drastic consequences for the well-being of the members.
The German Research Foundation (DFG) is funding this research by the Collaborative Research Center (CRC/SFB 1182) entitled “Origin and Function of Metaorganisms”. The overall goal of the CRC 1182 is to improve understanding the role of multi-organismic interactions for health and disease. We are particularly interested in the specific functional consequences of the interactions, the underlying regulatory principles, and also the resulting impact on host life history and evolutionary fitness in selected host systems.
Recent news
We communicate all our observations and results directly to the public in our news section.